16 Uncovering the Source
⬜ Developing Hypotheses
⬜ Sample Collection
⬜ Outbreak Investigation
🟩 Sequencing
🟩 Bioinformatics
⬜ Molecular Epidemiology
⬜ Public Health Implementation
16.1 Testing the muubats
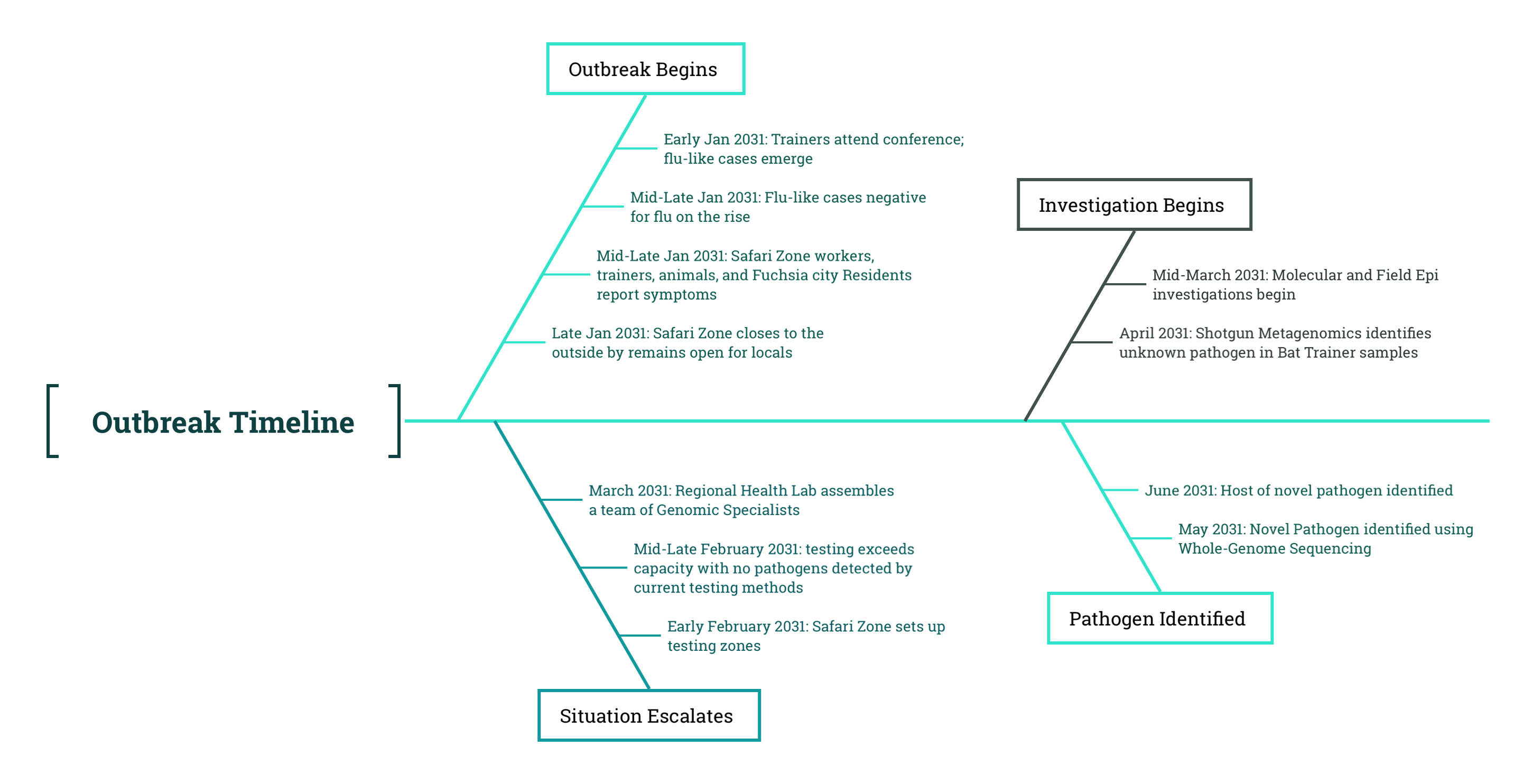
New swab samples were collected from the muubats, and samples were initially tested using a PCR assay developed specifically for the novel virus by the Regional Health Lab. The PCR assay targeted the S and N genes based on the consensus genome taken from one of the bat trainers. However, and to everyone’s surprise, the PCR results for the novel virus S gene and N gene came back negative, suggesting that the muubats did not currently harbor the novel virus. At first, this seemed like a dead end. If the muubats weren’t carrying the virus now, how could they have been the source? But then someone remembered: All newly introduced animals had archived samples from their initial intake exams. Be that as it may, the team remained unconvinced considering new evidence, and continued to bounce hypotheses back and forth.
Amid the discussion, someone asked, “isn’t it protocol when testing new animal species to keep a set of samples stored for potential future uses? I think we should test the first swab samples obtained from the muubats back in December”. Given the uncertainty around the origins of the outbreak, the team decided to revisit earlier samples collected during routine testing and surveillance. Among them were the archived swabs from the muubats’ intake exams. If any clues had been overlooked, these samples might fill in the gaps.
With the whole team in agreement, the lab pushed forth on running a PCR test on the initial swab samples taken from the muubats. The PCR results came back positive for the S and N genes. The samples proceeded to RNA extraction and preparation for sequencing, and the samples were sequenced.
After a week, the results finally came back indicating that the novel virus was identified in the muubat samples taken from December of the previous year. The discovery raised new questions. If the virus was present in the muubats months earlier, why was it not detected or causing illness at the time? What may have changed between the original exposure and the emergence of symptomatic cases in humans and animals? The multidisciplinary team met once again to deliberate.
16.2 Testing and Sequencing of Human and Animal Samples
“Now that we have a suspect pathogen,” a microbiologist stood up to speak, “We should run the same PCR assay on the human and animal samples. If the results return positive for the S and N genes of the novel virus, then we should sequence them for further analyses.”
Everyone was in agreement and quickly got to work. The Microbiologists ran the PCR assays on all samples, with plans to proceed to sequencing if the results were positive. If the sequencing run yielded high-quality raw reads, the Bioinformaticians would perform quality control and process the data using their in-house genome assembly pipeline.
Shortly after, the PCR assay results came back and all samples were positive for the S and N genes. The results were decisive, and the team felt the momentum shift — they were onto something.
The Microbiologists then sequenced the samples, and the sequencing run yielded a large volume of raw reads, short fragments of DNA and RNA sequences generated by the machine. But raw data alone wasn’t enough to move forward. Before any genome assembly could begin, the Bioinformaticians needed to perform a series of pre-genome assembly quality control (QC) steps to ensure the data was reliable.
16.3 Pre-Genome Assembly QC
One of the first things they examined was the read quality score for each base in the sequence. These scores, often referred to as Phred scores, indicate the probability that a particular base call is incorrect. A higher score means higher confidence. Reads with many low-quality bases (i.e.typically at the ends) are prone to errors and can introduce false variants during genome assembly. To mitigate this, the team removed or trimmed low-quality bases from the reads to retain only high-confidence data.
Next, the Microbiologists checked for and trimmed adapter sequences, short artificial DNA fragments that are added during the library preparation process so the sequencer can recognize and read the sample. If these adapters aren’t trimmed out, they can contaminate the data and interfere with downstream analyses, especially alignment and assembly. Specialized tools scan for known adapter sequences and remove them from the raw reads.
They also looked for overrepresented sequences and duplicate reads, which can suggest library preparation artifacts, contamination, or biased amplification. These patterns can distort coverage or make a dataset appear more robust than it actually is. Removing redundancy helps ensure that the reads truly reflect the diversity of genetic material in the original sample.
Another important step involved assessing GC content, the proportion of guanine (G) and cytosine (C) bases in the sequences. Abnormal GC content compared to what’s expected for the target organism can suggest contamination or poor amplification. GC bonds typically require more heat to break than AT bonds because they contain more hydrogen bonds than AT, which can further complicate wet lab methods. The Bioinformaticians also screened for reads that aligned to common laboratory contaminants or host genomes, excluding these from the dataset to focus on relevant microbial content.
After passing all of these checks, the data was considered high quality. With confidence in their sequencing reads, the team proceeded to genome assembly — now working from a dataset that was clean, trimmed, and ready to be pieced together.
Summary of Bioinformatics Pre-Genome Assembly QC
| QC Step | Why It Matters | How It’s Typically Done (Tool-Agnostic Description) |
|---|---|---|
| Read Quality Scoring | Identifies bases with high error probabilities that can cause false variants. | Base-level quality scores are used to trim or filter out low-confidence regions. |
| Adapter Trimming | Removes artificial sequences that interfere with alignment or assembly. | Reads are scanned for known adapter patterns and trimmed accordingly. |
| Overrepresented Sequence Check | Detects artifacts or biased amplification that distort microbial composition. | Repeated sequences are flagged as disproportionately frequent and optionally removed. |
| Duplicate Read Removal | Reduces artificial inflation of coverage caused by PCR duplicates. | Identical or highly similar reads are collapsed into single representative instances. |
| GC Content Assessment | Flags contamination or poor amplification based on sequence composition. | GC content is compared to expected profiles for the target organism. |
| Contaminant Screening | Ensures non-target DNA (e.g., human or lab contaminants) is excluded. | Reads are mapped against reference genomes to filter out known contaminant sequences. |
“This is it! We got our sequencing data!” one of the Bioinformaticians exclaimed.
“Great work you guys! I can’t believe how smoothly everything went, it’s almost unnatural!” applauded the Molecular Epidemiologist.
The team cheered — it felt like their first real win. Excitement rippled through the room, and the momentum was palpable. They had cleared the first major hurdle. But they all knew: the hardest questions were still ahead.
16.4 Discussion Nine: Understanding and Working with New Evidence
A. What are the possible implications of this new piece of evidence? How might it change your understanding of the outbreak timeline or transmission dynamics?
B. Based on all the information gathered so far, what is a plausible hypothesis about the origin or source of the novel virus?
C. What genomic analyses would you propose to test your hypothesis? Consider what types of comparisons or visualizations would strengthen your conclusions.